今天,分享一篇分子之心创始人许锦波AI蛋白质设计最新进展,希望以下分子之心创始人许锦波AI蛋白质设计最新进展的内容对您有用。
编辑 | 绿萝
1 月 11 日,在机器之心 AI 科技年会上,分子之心创始人、美国芝加哥丰田计算技术研究所终身教授、清华大学智能产业研究院(AIR)卓越访问教授许锦波发表主题演讲《AI 蛋白质设计最新进展》,在演讲中,他介绍了蛋白质结构预测与蛋白质设计,他表示 AI 蛋白质结构预测只是一个开始,分享了分子之心开发的 AI 蛋白优化和设计平台——MoleculeOS,以及在蛋白质侧链、抗体抗原复合物结构预测的最新研究成果。「人工智能颠覆了蛋白质结构预测,并正在改变蛋白质优化设计。」
以下为许锦波在机器之心 AI 科技年会上的演讲内容,ScienceAI进行了不改变原意的编辑、整理:

大家好,我是许锦波。非常荣幸能够在机器之心 AI 科技年会上和大家讲讲我最近在 AI 蛋白质方面的研究进展。
蛋白质与蛋白质结构预测
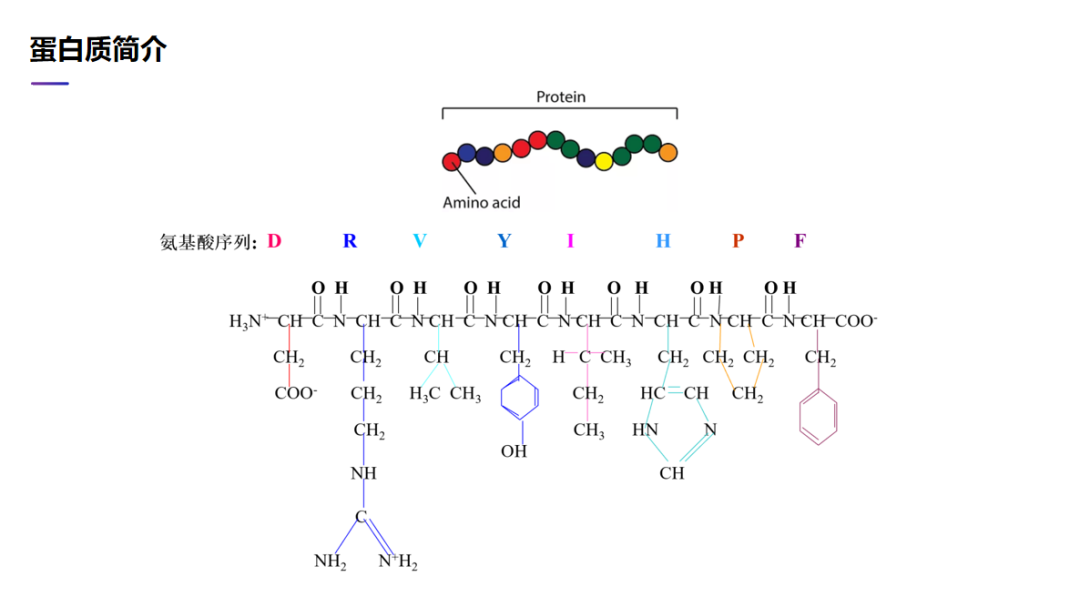
首先,我简单介绍一下蛋白质。蛋白质是一个非常大的分子,由 20 种氨基酸通过化学键串联在一起。在这里看一下蛋白质的分子式。这是一个很小的蛋白质,只有 8 个氨基酸。每个氨基酸都是由很多原子构成的,它有主链的原子,也有侧链的原子。自然界有 20 种氨基酸,它们的主链原子构成都是一样的,只是在侧链原子构成上不一样。有些氨基酸有很多侧链原子,有些氨基酸几乎没有侧链原子。这些原子在细胞里面相互作用,最后形成一个稳定的状态。
这里看一些蛋白质结构的例子,也就是,蛋白质在细胞中,它们的原子堆叠之后,最后的形状是什么样子的。这里一共有 4 个例子。假设我们已经有了蛋白质原子的相应的位置,现在可以用一些软件,可以把蛋白质的构象显示出来。比如上面两个图是用卡通图来表现蛋白质结构,很漂亮。下面把它们的原子也都给显示出来了,这个蛋白的结构看起来就比较复杂。
蛋白质的结构,特别是在原子层面的结构,对于理解蛋白质的功能非常有用。我们怎样才能得到蛋白质的结构?在过去几十年,科学家们开发了不同的实验技术,有三种主流的实验技术:晶体衍射,核磁共振以及冷冻电镜。这三种实验技术都可以把蛋白质的构象测出来,但是它们都存在一些问题,首先它们测一个蛋白结构都需要很长时间,可能耗时几个月到几年,花费也非常高。并且有很多蛋白质结构很难用这些实验技术给测出来。
AI 蛋白质结构预测,这只是一个开始
所以,另一种思路,就是我们利用计算的方法把蛋白质结构预测出来。现在,我们有高通量测序,可以很容易得到蛋白质的氨基酸序列。所以,假设我们有了蛋白质的氨基酸序列,能不能利用计算的方法把蛋白质的所有原子的三维坐标给计算出来,也就是蛋白结构预测。当然,这个问题是个很难的问题,已经研究了五六十年,一直没有很好的结果。直到最近几年,通过使用人工智能的方法,我们在蛋白质结构预测方面取得了很大的突破。人工智能预测蛋白质结构,在 2020 年和 2021 年都被《科学》杂志评为十大科学突破。去年 2022 年也入选了《麻省理工科技评论》的十大突破性技术。
从这里可以看出,蛋白质结构预测是个很难的问题,本身也是非常重要的问题,所以受到学术界的高度关注。AI 预测蛋白质结构取得了很大成功,但这只是一个开始,也只是用 AI 去研究蛋白质的一个开始。AI 蛋白质结构预测改变了分子生物学家的研究范式。以前大家都是基于蛋白质的氨基酸序列来研究一个蛋白质结构,但现在我们可以很容易地预测蛋白质结构。所以很多分子生物学家现在都可以基于蛋白质结构去研究蛋白质的功能。从另外一个角度,AI 预测蛋白质结构的成功,其实也证明了 AI 对蛋白质分子的研究是非常有效的。
AlphaFold2(AF2)可以把单一蛋白结构预测得很好,也有做得不是很好的地方,比如预测两个或多个蛋白质形成的复合物结构。虽然 AF2 在目前的方法里算是不错的,但是它的成功率还是远远不能让你满意的。特别是在一个非常重要的应用——抗体抗原相互作用预测方面,AF2 结果还是不那么好。当然也有很多 AF2 本身是不能做的,比如 AF2 没办法对蛋白质进行优化和重头设计,也没法预测单点突变对蛋白质结构和功能的影响。另外,AF2 可以预测蛋白质与蛋白质的相互作用,但是它没办法预测蛋白质和其他分子(小分子、DNA 和 RNA)的相互作用。

MoleculeOS:AI 蛋白优化和设计平台
蛋白质优化设计是创造自然界中不存在的蛋白质,即找到一个结构和功能可满足特定需求的氨基酸序列。蛋白质优化设计是一个非常难的问题,它面临的挑战,首先就是蛋白质序列空间是非常巨大的。假设我们只考虑一个 100 个氨基酸的蛋白质,自然界有 20 种氨基酸,其实序列空间就是 20 的 100 次方,这是个非常巨大的一个序列空间。但是巨大的序列空间里面其实只有很小比例的氨基酸序列可以折叠成一个稳定的构象,并且具有某种特定的功能。所以我们要在巨大的空间里面搜索,可以折叠成一个稳定的构象且具有我们所需要功能的氨基酸系列。这相当于大海捞针,是一个非常困难的问题。预测蛋白质结构和功能是蛋白质设计的一个基础,也就是我们在蛋白质设计的时候,往往需要对蛋白质结构和功能进行预测。
蛋白质设计在诊断、治疗和预防里面都有非常重要的应用。比如我们可以设计一个小蛋白,它可以用来做新冠病毒的诊断。当然我们也可以设计疫苗,用来预防新冠病毒。也可以用蛋白设计的方法去设计一个小蛋白,功能有点类似于抗体,它可以阻断新冠病毒进入人体。
分子之心在过去一段时间里,专门开发了一个 AI 蛋白优化和设计平台——MoleculeOS。这个平台主要是用来发明而不是发现蛋白质,实现「可编程」的蛋白质疗法。这个平台一共分为 4 层,在基础层上,是我们的一些 AI 算法,以及一些大数据库,还有一些算力平台。在能力层上,包含了不同的功能模块。比如我们可以做单个蛋白质预测,也可以用来做蛋白质复合物预测,当然也可以预测蛋白质功能,还有单点突变对蛋白质功能和结构的影响,以及蛋白质从头设计。在应用层上,我们会把这种功能模块用到各种不同模态的蛋白质上,比如我们可以把它用在酶和抗体上。最后产品层,我们就利用 MoleculeOS 设计不同的,满足我们所需要的蛋白质,用来做大分子药物研究或者合成生物学。
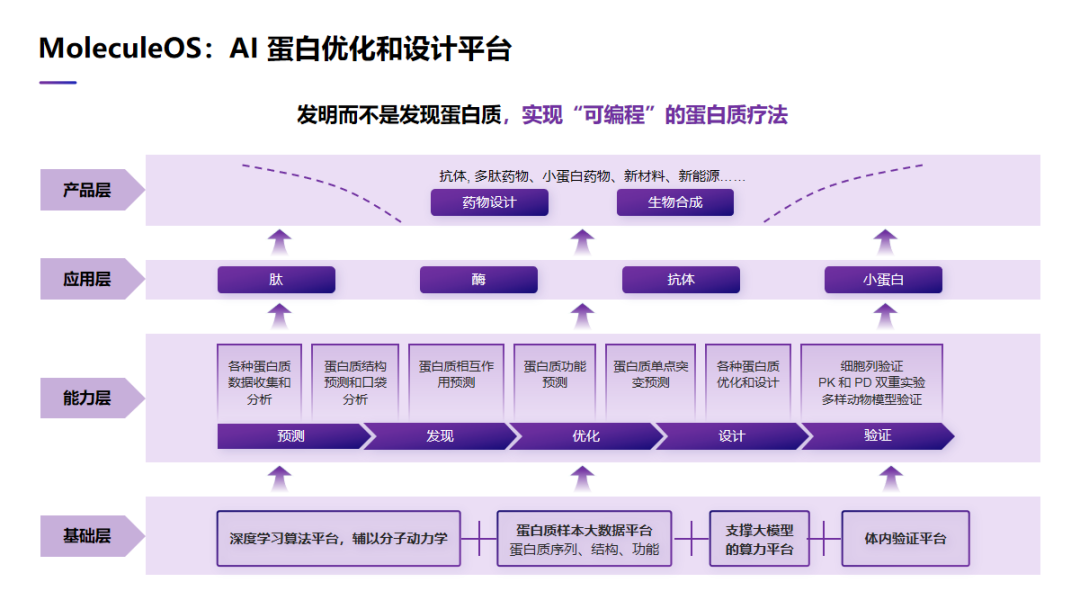
MoleculeOS 的底层是由不同的算法构成的。这里就列出了我们过去开发的 10 项全球领先的一些算法。比如我们有全球领先的蛋白质序列设计算法,也可以做非常复杂结构的蛋白质主链结构从头设计,也具有非常好的蛋白质侧链预测算法,以及蛋白质复合物,特别是抗体抗原结合的预测算法。下面讲一下具体的研究结果。

MoleculeOS 可以用来设计大小和构象不受限制的蛋白质。传统的蛋白质设计算法设计的蛋白质往往是比较小的,并且大多数情况下设计的蛋白质都是 Alpha 蛋白。但通过使用 AI 的方法,我们可以设计比较大的蛋白质,并且设计出来蛋白质的二级结构。它可以既有Alpha-helix,也有 Beta-strand,也就是我们可以设计的蛋白质空间比传统方法是要大得多。这也意味着潜在的可能性——我们可以设计蛋白质的功能也会比以前的方法更具有多样性。
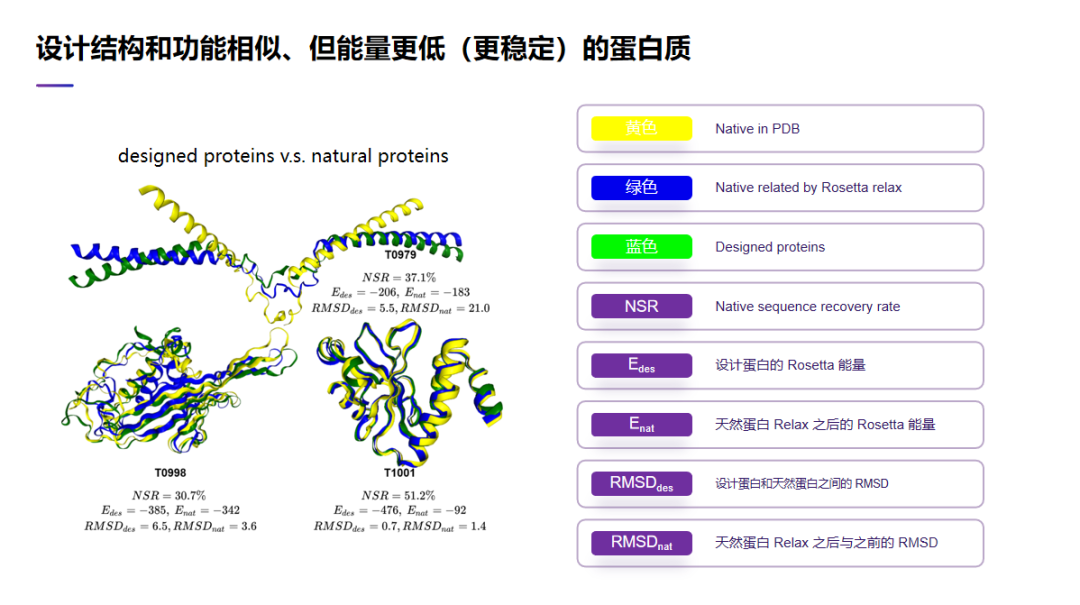
另外我们也可以设计结构和功能相似但是能量更低、更稳定的蛋白质。这里看 3 个例子。在这 3 个例子里,黄色的是实验结构,蓝色的我们设计出来的蛋白质。从这里可以看到,我们设计的蛋白质其实是跟实验结构看起来差不多,但如果计算它们的能量,我们设计出来蛋白质的能量往往是要低于自然界存在的蛋白质的能量。
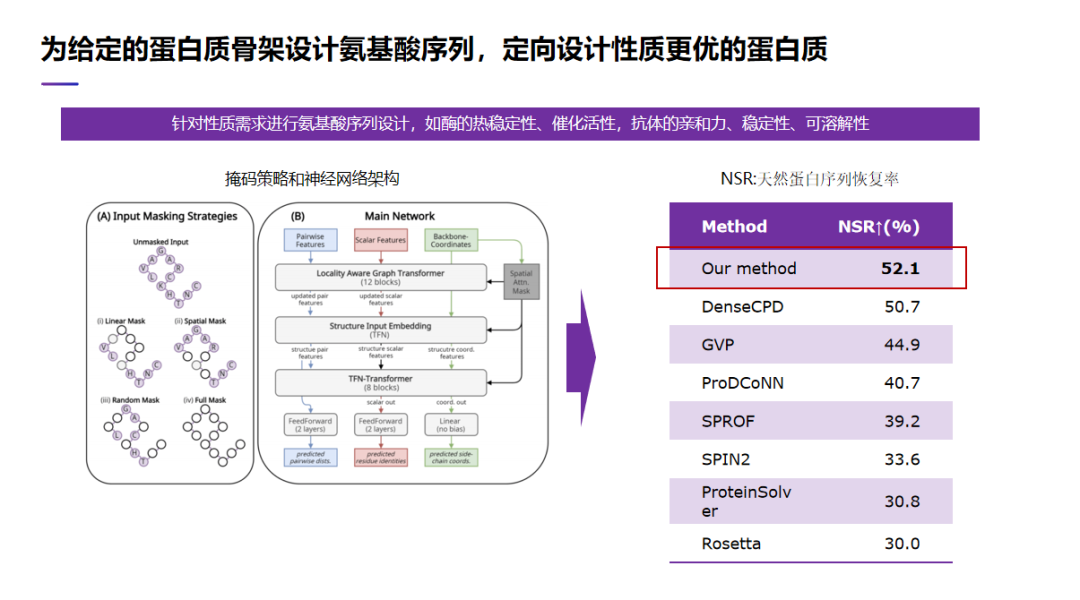
有时候给定一个蛋白质骨架,我们想重新设计蛋白质的氨基酸序列,就可以对蛋白质进行优化。比如我们可以针对性质需求进行氨基酸序列设计,如酶的热稳定性、催化活性,抗体的亲和力、稳定性、可溶解性。我们设计了一个 AI 算法,下图左边画出了我们 AI 算法的神经网络架构,右边表里面列出了性能评估。我们可以有各种方法去评估氨基酸序列设计算法,在这里我们用一种通用的评估标准,也就是计算天然蛋白序列的恢复率。与其他的方法相比,我们得到恢复率是最高的。
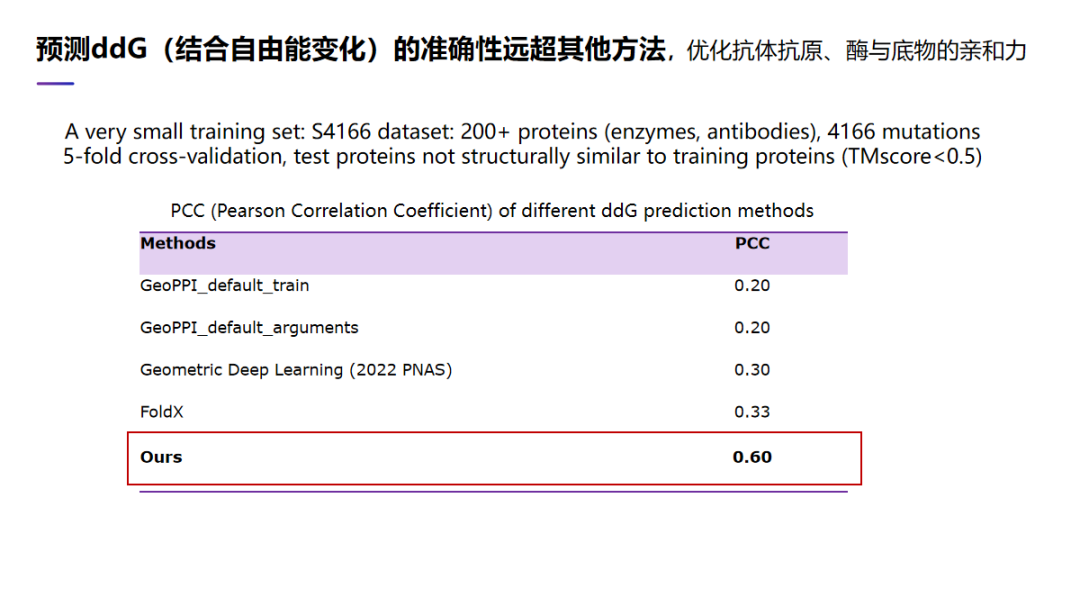
另外一个非常重要的功能模块,就是预测两个或多个蛋白质结合自由能的变化,即 ddG 预测。ddG 预测可以用来做抗体优化、酶与底物的亲和力的优化、以及用来优化某个蛋白质,比如说多肽分子。这个问题难度在于,我们并没有很多数据可以用来训练我们的 AI 模型。在这种情况下,往往很多 AI 算法是可很容易造成了overfit。在这里我们设计了一个很好的算法,我们测量算法预测的结果跟实验结果的相关性,也就是 PCC。我们的算法的 PCC 可达到大概 60%,远远好于这以前的方法,比如 FoldX,还有一些深度学习方法。不管以前方法是传统的方法还是深度学习方法,他们的 PCC 都远低于我们。
研究成果:蛋白质侧链、抗体抗原复合物结构预测
接下来,更详细地讲讲我们最近的一些结果。
在蛋白质侧链结构预测上,我们取得了比较好的进展。所谓的蛋白质侧链结构预测,也就是假设我们已经有了蛋白质的主链接结构,我们可以通过预测把侧链的原子位置确定下来。传统的方法主要是使用能量优化。首先,因为侧链原子在空间的分布不是随机的,他们需要对侧链原子在空间分布进行做 cluster,把它分成不同的组。然后再针对某一个特定的氨基酸去搜索它的侧链分组,同时优化能量。这是传统的方法。最近我们设计了一个 AI 算法,用来预测蛋白质侧链结构。
这张表里面展示一些结果。首先我们在 CASP13 这些测试蛋白上面测试了我们算法,使用的主链结构是实验结构。把我们的方法跟其他方法比较,有三种传统的方法, RosettaPacker, SCWRL 和 FASPR,也有深度学习方法,比如 DLPacker。我们这里使用了两种策略。从这里可以看到,我们的方法无论使用哪一种测量指标,都是要好于以前的方法。比如 RMSD,就是我们预测出原子的位置与它实验结构测出的位置的误差——当然误差是越小越好——从这里可以看到,我们两种方法得到的 RMSD 都是有远低于以前的四种方法的。在二面角预测的误差上面也是一样的。在二面角预测误差上面,我们的结果也是要好于以前的 4 种方法,特别是在第一个二面角上面,我们的结果是远好于以前的 4 种方法。
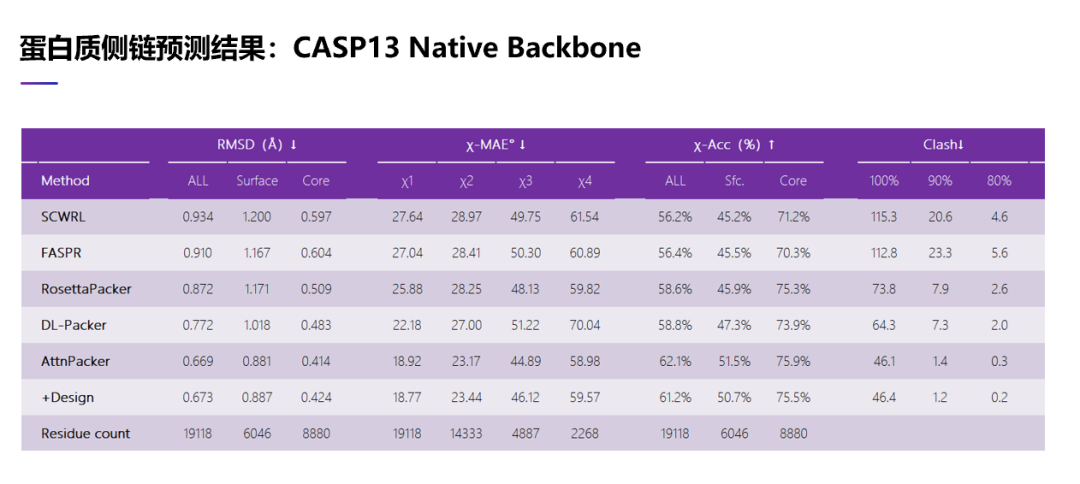
另外我们也检查了我们预测到的侧链原子在空间中是不是有冲突,也就是它在物理上是不是可行的。其实从表里面可以看到以前的 4 种方法,它们预测出来的原子在空间中都有很多冲突,在物理上不一定可行。我们的预测出来侧链原子位置,它的冲突比以前的方法要少很多。我们的冲突数目其实接近实验结构解出来的蛋白质结构里包含的冲突数目。
我们也在 CASP 14 上做了测试,主链结构同样使用了实验结构,我们也将我们这两种策略与以前的 4 种方法在数据集上进行了比较,可以看到还是一样的结果。比如我们做出了侧链原子结构预测,无论是使用 RMSD 评估也好,还是用二面角误差评估,或者用冲突的数目评估我们的算法性能,都是远好于以前的方法。特别是在冲突的数目指标上,我们预测的侧链原子位置甚至都要好于用实验结构解出来的侧链原子的位置,可以用我们这个方法去优化一个用实验结构解出来的蛋白质结构。
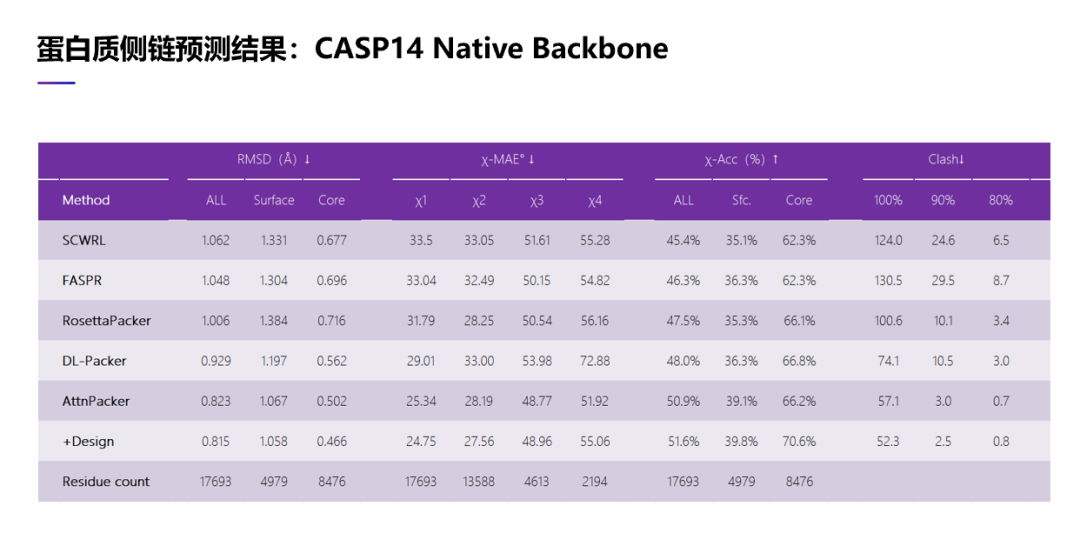
另外,我们也看了 56 个在 CASP 13 和 CASP 14 里比较难的那些测试例子,在测试里面,我们主链结构同样使用了实验技术做出来的结构。在数据集上,结果与前面两个数据上结果是一样的。无论在 RMSD 评估上,还是二面角误差评估,或者在冲突的数目上。在冲突数目上,我们预测出来的侧链原子冲突数目甚至要少于用实验结构解出来的冲突数目。
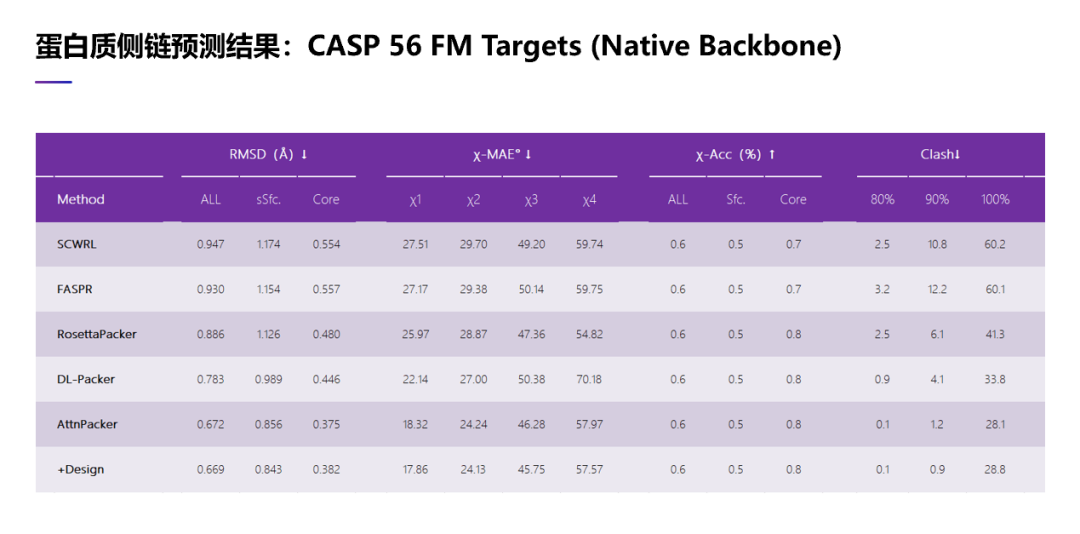
除了用实验技术解出来的主链结构作为输入,我们也考虑了其他情况。比如,假设主链结构是用预测出来的,而不是用实验技术解出来的,看看我们算法的效果怎么样。在这里,我们测试了 CASP 13 的蛋白,但是在这个测试例子里,主链结构不是实验技术解出来的,而是预测出来的。在这里,我们使用了 AF2 加上模板,再加上多序列比对去预测主链结构。并且我们只考虑那些 AF2 预测出来效果比较好的例子,也就是 RMSD 小于 2. 5 的测试蛋白,一共有 47 个。除了跟以前的四种侧链预测方法做比较,我们也比较了另外三种预测方法,比如我们比较了 AF2 两种不同的策略,一个是使用了多序列比对,另外是没有使用多序列比对。另外我们也比较 了 OmegaFold。
从这里可以看出,我们的方法是有最小的三维坐标的预测误差,即最小的 RMSD。从二面角来说,我们也是最小的二面角误差。考虑预测出的原子在空间中的冲突,同样我们预测出来原子在空间中冲突要远远好于其他的方法,就是我们预测出来侧链原子在空间中的冲突数目非常接近用实验技术解出来的结构里原子在空间中的冲突数目。
我们也看了 CASP 14 的结果,在这里,同样我们只考虑那些实验法预测的比较好的那些 CASP 的测试蛋白。与 CASP 13 的结果趋势是非常相似的。当只考虑是 RMSD 时,我们的结果要好于其它方法。在二面角误差上面,也是比 AF2 好一些。在原子的冲突数目上,我们预测出原子冲突数目非常接近用实验技术解出来的,要好于 AF2 预测出来的结果。
当我们考虑所有的 CASP 13 测试蛋白,在这种情况下,我们也考虑那些主链预测得不是很好的情况,看看那些测试蛋白效果怎么样。其实这个趋势是与以前一样,也就是我们预测出来侧链原子的位置。无论是 RMSD,还是二面角误差,还是原子的冲突数目,我们的结果都是最好的。
同样我们也考虑了 CASP 14。我们观察到一样的趋势,也就是,我们预测效果是要好于以前的方法,也好于 AF2。特别是在原子冲突数目方面,我们预测的结果甚至要好于实验技术解出来的结构。
刚才讲了我们最近在蛋白质侧链预测上的结果。下面讲一下我们在抗体抗原形成的复合物结构预测上的效果。这是另外一个 AI 算法。我们的 AI 算法在抗体抗原复合物结构预测上取得了比较大的进展。首先我们在这里测试了两种情况。第一种情况是,假设我们知道单个抗体或单个抗原的实验结构,我们怎么能够预测抗体和抗原结合在一起的结构?我们想看看在这种情况下我们预测出来的效果怎么样。在这里我们也用了几不同的指标去评价预测出的结果。比如我们也使用了 DockQ 的成功率,这个指标是越高越好。除了 DockQ 成功率之外,我们也计算了 I-RMSD,也就是抗体抗原结合的那些位置的预测误差,所以这个指标越小越好。除了 I-RMSD,我们也计算了 L-RMSD。在计算 I-RMSD 和 L-RMSD 时,我们考虑了比如前 25%,50%,75% 的预测结果误差。我们的算法可以生成多个不同的复合物构象。在这里除了衡量,然后我们会用一个预测 pLDDT 去排序。所以选出前 5 个预测出来。除了评估第一个预测,我们也评估了前 5 个里面最好的复合物结构的质量是怎样的。当然有些算法他们只能生成单个,或者他们虽然能生成多个预测结构,但是可能比如他们可以生成 5 个,但是 5 个可能预测的结构其实也是比较相似的。在这里我们评价了第一个预测结构和前 5 个里面最好的预测结构。
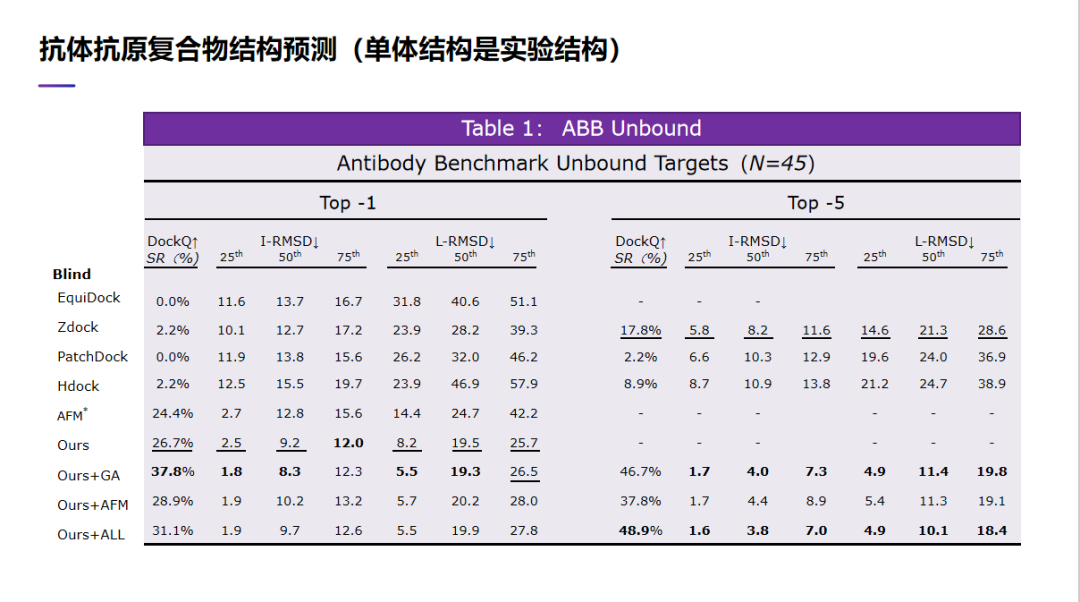
从这张表里面可以看到。首先,传统的一些蛋白质对接算法,比如 Zdock,PatchDock,Hdock,甚至最近一个基于深度学习的 EquiDock,他们的 DockQ 成功率其实都是非常低的,都是个位数。他们预测出的 I-RMSD 和 L-RMSD 也都非常大。我们也测试了 AlphaFold-multimer(AFM*),DockQ 的成功率大概是 24.4%,这个结果是远远好于传统的一些蛋白质对接算法。
对于我们的方法,我们试了 4 种不同的策略。对于每个测试的复合物,如果我们只考虑第一个预测结构,我们的成功率大概是 37. 8%,远远好于 AFM* 。我们也看了 I-RMSD 和 L-RMSD,无论是 I-RMSD 还是 L-RMSD,我们的误差值都是远小于 AFM*。我们也看了前五个预测出来结果,在前五个预测结果,我们成功率就变成了 48. 9%。这个成功率是也好于我们只考虑第一个的成功率,也就是,我们预测出来的这些复合物结构,它其实是有多样性的。这样,如果我们考虑前 5 个预测结果,我们可以大幅度提高预测的成功率。
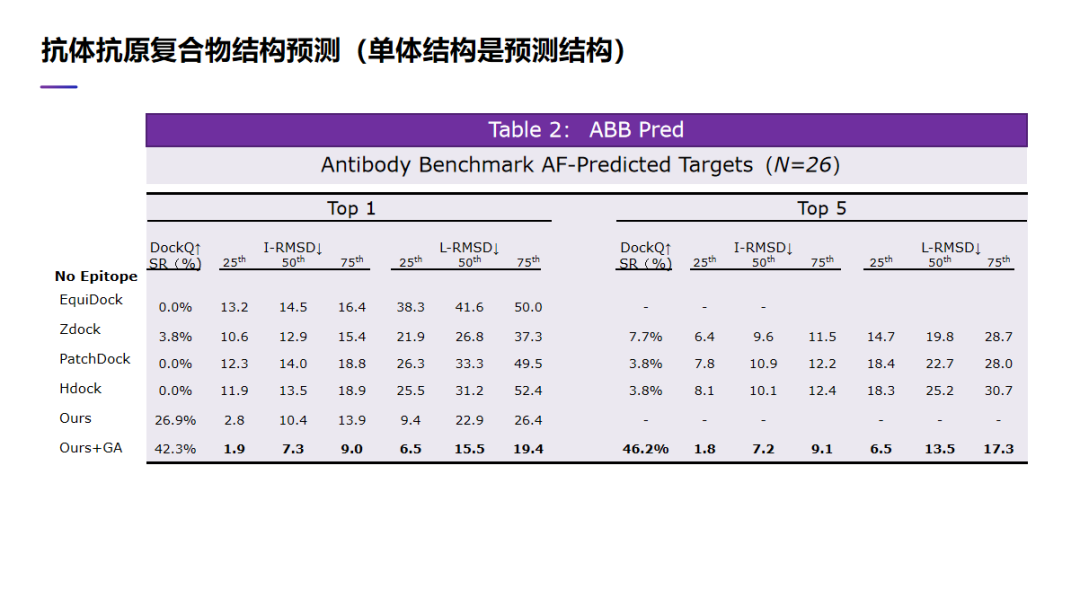
刚才是假设我们有抗体或者抗原单体的实验结构,但在某些情况下,我们是没有它的实验结构,我们只有单体的预测结构。这时,我们的算法到底能在预测结构的情况下,能做得有多好。同样,我们比较了我们的方法,与一些传统的蛋白质对接算法,比如 Zdock,也比较了最近的一个深度学习算法:EquiDock。同样,我们的成功率是远远好于其他的蛋白质对接方法。比如我们如果只考虑前第一个预测复合物结构,我们的 DockQ 成功率是大概 42. 3%。如果考虑 5 个里面最好的,我们的 DockQ 成功率是 46. 2%。
所以,综合这两个表,可以看到,无论是使用单体的实验结构,还是使用单体的预测结构,我们的抗体抗原复合物结构预测算法的性能是要远好于其他的方法。

最后,我总结一下。人工智能不但颠覆了蛋白质结构预测,其实也大幅度地提高了蛋白质其他方面的研究的性能。比如在蛋白质侧链结构预测,在蛋白质复合物,特别是抗体抗原复合物结构预测方面,在蛋白质的优化和设计方面,通过使用人工智能,我们都可以做得比传统方法要好很多。蛋白质的优化和设计有非常广阔的应用场景。它在制药、合成生物学、工农业生产、材料设计以及环境改善方面都有非常广阔的应用场景。