原标题:“史上最严”医疗器械监管条例来了,变化有哪些?
6月1日起实施的新修订的《医疗器械监督管理条例》(以下简称“《条例》”)堪称史上最严。
与2017年修订版相比,新《条例》从原来的8章80条修订、增补为8章107条,在落实注册人制度、鼓励创新、引入医疗器械“电子身份证”、严重违法者“终身禁业”等方面均有新表述。
在近日召开的首届“长三角医疗器械高质量发展高峰论坛”上,国家药品监督管理局医疗器械注册司稽查专员王兰明从监管视角对新《条例》进行了详细解读。
焦点一:新增医疗器械注册人制度
王兰明介绍,新《条例》新增了医疗器械注册人制度。
据新华社消息,医疗器械注册人制度是国际上普遍采用的现代医疗器械管理制度,也是新《条例》修订的核心制度之一。
根据新《条例》,国家对医疗器械按照风险程度实行分类管理,第一类医疗器械风险程度低,第二类具有中度风险,第三类具有较高风险。对第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。
新《条例》第八章第103条第2款规定,医疗器械注册人、备案人,是指取得医疗器械注册证或者办理医疗器械备案的企业或者研制机构。
该条规定最大的变化是什么?王兰明解读道,最大的变化就是引入研制机构作为注册人、备案人,注册人、备案人不再局限于生产企业。
新《条例》第三章第34条规定,医疗器械注册人、备案人可以自行生产医疗器械,也可以委托符合本条例规定、具备相应条件的企业生产医疗器械。
浙江省药监局医疗器械监督管理处处长戴桂平在接受澎湃新闻(www.thepaper.cn)采访时表示,实施注册证持有者和产品生产者分离,这使得科研机构和科研院所能专注创新,把生产交给专业厂商,从而促进科研成果快速转化,推动创新高质量发展。
同时,新《条例》第二章第13条规定,医疗器械注册人、备案人应当加强医疗器械全生命周期质量管理,对研制、生产、经营、使用全过程中医疗器械的安全性、有效性依法承担责任。
王兰明表示,注册人制度的核心意义,就是构建一个贯穿医疗器械全生命周期的责任主体,由该责任主体承担起医疗器械质量从始至终的责任,在一定条件下,产品注册证和生产许可证可以解绑。
关于注册人制度的目标,王兰明表示,希望能够以此鼓励技术创新、优化资源配置、让专业的人干专业的事、压实主体责任、提高管理水平,最终实现激发医疗器械市场红利的目标。
事实上,我国医疗器械注册人制度试点工作已于2018年起步,当年,上海市、广东省、天津市三地先后开展试点。
及至2019年8月,国家药监局发布《关于扩大医疗器械注册人制度试点工作的通知》,医疗器械注册人制度试点扩大至北京、河北、黑龙江、江苏、浙江、湖南、重庆、福建等21个省、自治区、直辖市。此次新《条例》的实施,则使医疗器械注册人制度在全国范围内落地。
焦点二:新增鼓励和支持医疗器械创新的规定
王兰明介绍,新《条例》新增了鼓励和支持医疗器械创新的相关规定。
新《条例》第一章第8条规定,国家制定医疗器械产业规划和政策,将医疗器械创新纳入发展重点,对创新医疗器械予以优先审评审批,支持创新医疗器械临床推广和使用,推动医疗器械产业高质量发展。
王兰明介绍,创新医疗器械审批程序始于2014年。国家药监局官网显示,原国家食品药品监督管理总局制定的《创新医疗器械特别审批程序(试行)》于2014年3月1日起实施,并于2018年进行了修订。
新修订的程序对具有我国发明专利、技术上具有国内首创、国际领先水平并且具有显著临床应用价值的医疗器械设置了特别审批通道。对于经审查同意按该程序审批的创新医疗器械,国家药监局及相关技术机构,按照“早期介入、专人负责、科学审查”的原则,在标准不降低、程序不减少的前提下,予以优先办理。
截至2021年5月底,国家药监局已批准91个公司109个创新医疗器械上市,其中包括基因测序仪(深圳华因康基因科技有限公司生产,2014年批准);骨科手术导航定位系统(北京天智航医疗科技股份有限公司生产,2016年批准);无创血糖仪【博邦芳舟医疗科技(北京)有限公司生产,2019年批准】;以及2021年4月批准的临时起搏器(深圳市先健心康医疗电子有限公司生产),据澎湃新闻此前报道,该产品是国产首例临时心脏起搏器。
此外,原国家食药监局组织制定的《医疗器械优先审批程序》已于2017年1月1日起实施,对诊断或者治疗罕见病且具有明显临床优势、诊断或者治疗恶性肿瘤且具有明显临床优势、列入国家科技重大专项等情形的医疗器械注册申请实施优先审批。
王兰明介绍,上述优先审批措施发布以来得到了业内的肯定,这也给予监管部门诸多鼓励。
新《条例》的第一章第9条还规定,国家完善医疗器械创新体系,支持医疗器械的基础研究和应用研究,促进医疗器械新技术的推广和应用,在科技立项、融资、信贷、招标采购、医疗保险等方面予以支持。
王兰明表示,该条规定较为严格,更具产业化,“我相信条例实施后,各个部门都会抓紧落实,在各个方面和药监部门一起来推动医疗器械技术创新工作。”
焦点三:我国患者有望用上全球最新创新医疗器械
王兰明表示,新《条例》新增进口创新医疗器械注册、备案可以免于提交境外上市许可证明文件。
新《条例》第二章第15条第二款规定,(第一类医疗器械产品备案)未在境外上市的创新医疗器械,可以不提交备案人所在国(地区)主管部门准许该医疗器械上市销售的证明文件;第二章第16条第二款规定,(申请第二类、第三类医疗器械产品注册)未在境外上市的创新医疗器械,可以不提交注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。
王兰明解读,从2000年至今,监管部门一直要求进口的医疗器械,要先取得境外的上市许可证明文件,到新《条例》,“创新医疗器械”可以免于提交相关文件。“这是为什么?这就是为中国的患者能得到最新的医学治疗,特别是对严重威胁人类生命的(疾病)。”
王兰明表示,这也意味着中国医疗器械监管到了一个新时期,将会有全球性的产品在中国首先上市,或者和其他国家(和地区)同步上市,为此,监管部门要做好相关准备。
此外,王兰明介绍,新《条例》还增加了医疗器械拓展性临床试验制度。
2020年3月发布的《医疗器械拓展性临床试验管理规定(试行)的公告》(以下简称“《公告》”)显示,医疗器械拓展性临床试验,是指患有危及生命且尚无有效治疗手段的疾病的患者,可在开展临床试验的机构内使用尚未批准上市的医疗器械的活动和过程。
新《条例》第二章第29条规定,对正在开展临床试验的用于治疗严重危及生命且尚无有效治疗手段的疾病的医疗器械,经医学观察可能使患者获益,经伦理审查、知情同意后,可以在开展医疗器械临床试验的机构内免费用于其他病情相同的患者,其安全性数据可以用于医疗器械注册申请。
王兰明表示,对此,上述《公告》已经对医疗器械拓展性临床试验提出了比较严格的要求,如对于试验机构,要求具有三级甲等医疗机构资质、已开展同种疾病研究和治疗、临床专业水平国内先进、具有与拟开展的拓展性临床试验相适应的诊疗项目;此外,要求开展医疗器械拓展性临床试验前,申办者应当向所在地省、自治区、直辖市药品监督管理部门备案等。
焦点四:引入医疗器械“电子身份证”
新《条例》下,每个医疗器械将有唯一的“电子身份证”供溯源管理。
王兰明介绍,新《条例》新增了医疗器械唯一标识制度。“医疗器械唯一标识在国际上叫UDI,这是一个国际通行制度,俗称医疗器械‘电子身份证’。”
2019年10月发布的《医疗器械唯一标识系统规则》显示,医疗器械唯一标识,是指在医疗器械产品或者包装上附载的,由数字、字母或者符号组成的代码,用于对医疗器械进行唯一性识别。医疗器械唯一标识系统,由医疗器械唯一标识、唯一标识数据载体和唯一标识数据库组成。
新《条例》第三章第38条规定,国家根据医疗器械产品类别,分步实施医疗器械唯一标识制度,实现医疗器械可追溯,具体办法由国务院药品监督管理部门会同国务院有关部门制定。
王兰明解读道,医疗器械唯一标识制度有利于加强医疗器械的追溯管理,有了这个像“身份证”一样的编码后,就能及时识别问题产品,实现迅速召回;同时,对打击假冒伪劣也有重要作用。
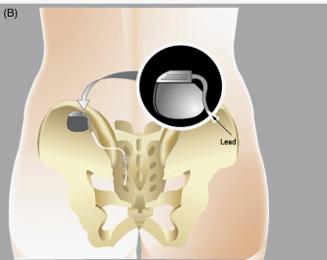
据国家药监局官网消息,医疗器械唯一标识系统试点工作自2019年7月开展,持续至2020年12月31日。试点结束后,2021年1月1日起,第一批实施医疗器械唯一标识工作全面启动,9大类69个品种的医疗器械纳入第一批实施。这69个品种包括可吸收缝合线、造影导管等手术器械;还有植入式心脏起搏器、植入式神经刺激器、髋关节假体、乳房植入物等植入器械。
此外,这69个品种中还包括《第一批国家高值医用耗材重点治理清单》中的耳内假体、脊柱椎体间固定/置换系统、可吸收外科止血材料、阴茎假体、植入式药物输注设备等5种高风险第三类医疗器械。
焦点五:史上最严医疗器械监管条例,新增“终身禁业”规定
王兰明介绍,新《条例》的一个特点,就是最严谨的标准、最严格的监管、最严厉的处罚。新《条例》的总体思路和主要修订内容之一,即是加大惩处力度,提高违法成本。
新《条例》第七章第81条规定,有“生产、经营未取得医疗器械注册证的第二类、第三类医疗器械”、“未经许可从事第二类、第三类医疗器械生产活动”、“未经许可从事第三类医疗器械经营活动”三种情形之一、情节严重的,责令停产停业,10年内不受理相关责任人以及单位提出的医疗器械许可申请,对违法单位的法定代表人、主要负责人、直接负责的主管人员和其他责任人员,没收违法行为发生期间自本单位所获收入,并处所获收入30%以上3倍以下罚款,终身禁止其从事医疗器械生产经营活动。
新《条例》第七章第83条规定,在申请医疗器械行政许可时提供虚假资料或者采取其他欺骗手段的,不予行政许可,已经取得行政许可的,由作出行政许可决定的部门撤销行政许可,没收违法所得、违法生产经营使用的医疗器械,10年内不受理相关责任人以及单位提出的医疗器械许可申请;情节严重的,责令停产停业,对违法单位的法定代表人、主要负责人、直接负责的主管人员和其他责任人员,没收违法行为发生期间自本单位所获收入,并处所获收入30%以上3倍以下罚款,终身禁止其从事医疗器械生产经营活动。
对比而言,在2017版条例中,并无终身禁止从业的相关规定,新《条例》堪称史上最严。





抖音、快手直播带货周报_00.png)